


 2021.11.22
2021.11.22  10155次
10155次
一、MAGeCK概要
MAGeCK是一种基于模型的全基因组 CRISPR-Cas9 敲除的分析计算工具,可从最近的基因组规模 CRISPR-Cas9 敲除筛选 (或 GeCKO) 技术中识别重要基因。由 Dana-Farber 癌症研究所刘晓乐博士实验室的 Wei Li 和 Han Xu 开发,并由国家儿童医学中心的 Wei Li 实验室积极更新。
二、MAGeCKFlute的主要功能
| 功能 | 描述 |
| mageck count | 将原始 FASTQ 数据映射到library参考文件并计算每个 sgRNA 的读数 |
| mageck test | MAGeCK RRA(通过计算 RRA富集分数 来识别 CRISPR 筛选中的hits以表明基因的重要性) |
| mageck mle | MAGeCK MLE(通过计算每个目标基因的“β分数”来衡量目标被扰乱后的选择程度,从而识别 CRISPR 筛选中的hits) |
| VISPR | MAGeCK 结果的可视化 |
| mageck-vispr | FASTQ 和原始计数级别的质量控制;包括所有的MAGeCK 和 VISPR 的功能 |
| BatchRemove | 消除CRISPR 筛选raw read-count水平的批次效应 |
| mageck test/mle, --cnv-normparameter | 更正使用 MAGeCK RRA 和 MAGeCK MLE 识别hits时由拷贝数引起的偏差 |
| mageck_nest.py | 提高hit识别率并移除outlier sgRNAs |
| FluteRRA | MAGeCK RRA结果的下游分析 |
| FluteMLE |
三、流程摘要
1、使用 MAGeCK 和 MAGeCK-VISPR 进行 CRISPR 筛选数据分析的基础知识
MAGeCKFlute 分别使用 mageck count 和 mageck test/mle 执行读取映射和命中识别,这是 MAGeCK 和 MAGeCK-VISPR 的主要功能(表 1)。MAGeCKFlute 的典型输入是 FASTQ 文件或原始read-count表,其中列是样本,行是 sgRNA。CRISPR 筛选分析通常包含两个部分:sgRNA 水平和基因水平分析。sgRNA水平分析模型独立读取单个 sgRNA 的计数,计算每个sgRNA的倍数变化和P值,类似于RNA-seq分析。基因水平分析整合了 sgRNA 水平倍数变化和 P 值,以识别感兴趣的基因hits。MAGeCK 首先将测序读数比对到 sgRNA 文库,然后标准化 sgRNA 读数计数以调整测序深度。
2、质量控制和read-count表生成
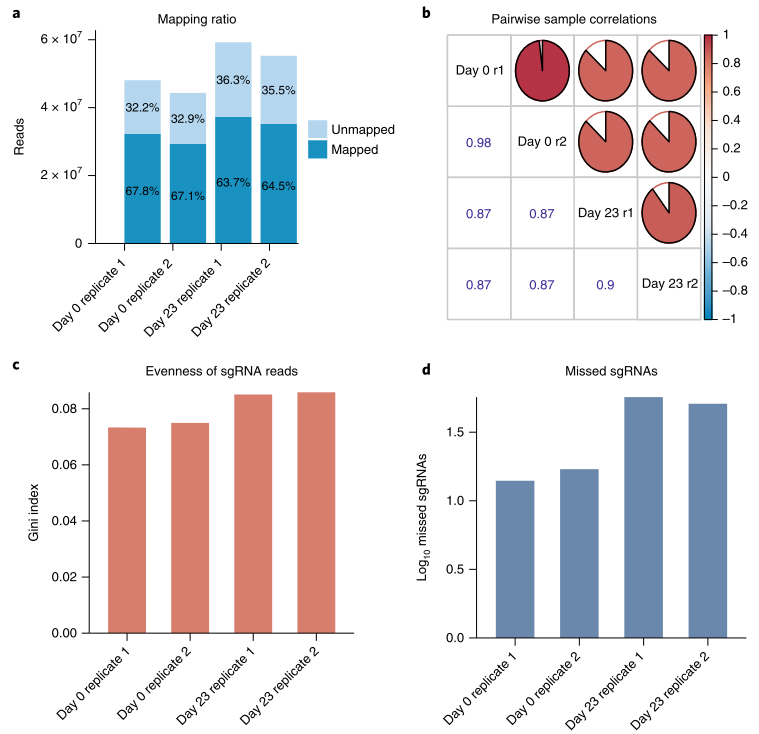
在识别hits之前,需要将reads与已知的 sgRNA 库对齐并评估筛选质量(图 2)。MAGeCK 和 MAGeCK-VISPR 将reads与 sgRNA 文库文件比对,计算每个 sgRNA 的read数量并输出一组 QC 统计信息,包括以下内容。
● 比对到的reads数量(图 2a);
● 比对到的reads的百分比(图 2a);
● 样本之间的read count相关性(图2b);
● Gini 指数(衡量 sgRNA read count的均匀性)(图 2c);
● 比对到0个reads的 sgRNA 的数量(图 2d)。
比对read百分比过低可能意味着存在寡核苷酸合成错误、测序错误或受污染的样品。高比对率则表明样品制备和测序成功。缺失sgRNA 量少也是高质量样本的良好指标。MAGeCK 和 MAGeCK-VISPR 使用基尼指数(这是经济学上衡量美国收入不平等的常用指标。)来衡量 sgRNA 读取计数的均匀性。高 Gini 指数表明 sgRNA read count在目标基因中分布不均匀。这可能是由 CRISPR 寡核苷酸合成的不均匀性、低质量的病毒文库包装、病毒转染效率低下或筛选过程中的过度选择引起的。
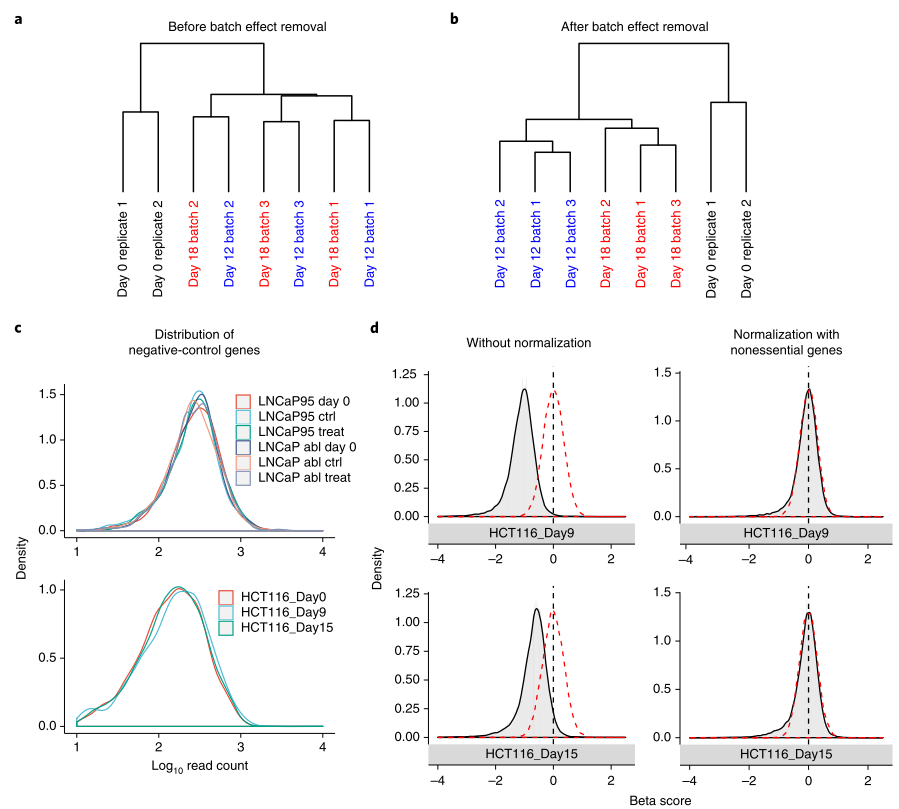
3、去除批次效应
在多个批次中执行或排序的 CRISPR 筛选可能包含批次效应。如果 CRISPR 筛选数据是使用不同的试剂或测序平台、在不同的时间或在实验条件的任何其他意外变化下生成的,则可以在这些数据中观察到批次效应。在这种情况下,去除批次效应是数据分析的必要步骤。一个例子是结肠癌中的公共数据库CRISPR 筛选,它具有很强的批次效应,其中样品按批次而不是按条件聚类(图 3a)。使用 ComBat 函数(合并到 MAGeCKFlute 中)在 sgRNA 计数级别校正数据集中的批次效应后,生物重复正确地聚集在一起(图 3b),这表明批次效应已被移除。
4、识别CRISPR 筛选hits
用于识别基因hits的第一种方法是 MAGeCK RRA。MAGeCK RRA 允许比较两个实验条件。它可以识别在两种条件下基本上选择的 sgRNA 和相应的基因。MAGeCK RRA 根据从负二项式模型计算出的 P 值对 sgRNA 进行排序,并使用名为 α-RRA 的修改后的 RRA 算法来识别正选择或负选择的基因。MAGeCK RRA 使用 RRA 富集分数来指示基因的重要性。可以模拟复杂实验设计的另一种方法是 MAGeCK MLE,它可用于分析来自具有多种试验条件的筛选数据,例如包含至少三个条件的典型药物筛选:第 0 天条件、对照条件(用媒介,如 DMSO)和药物治疗的条件。MAGeCK MLE 还模拟了 sgRNA 敲除效率,这可能因不同的序列内容和染色质结构而异。MAGeCK MLE计算每个目标基因的“beta score”,以测量基因扰动时的选择程度,类似于差异表达分析中的“对数倍数变化”测量。
MAGeCK-VISPR 进一步整合了 MAGeCK 的所有功能,并使用基于 Web 的交互式框架 VISPR 对所有结果进行质量控制和可视化。MAGeCK NEST 为 MAGeCK-VISPR 添加了功能以改进hit calling。首先,MAGeCK NEST 可以使用网络重要性评分工具 (NEST) 来整合来自蛋白质-蛋白质相互作用网络的信息,从而改善结果。其次,MAGeCK NEST 采用最大似然法去除 sgRNA 异常值,这些异常值通常具有更高的 G 核苷酸计数。如果有很多 sgRNA 异常值或如果在筛选数据中观察到高 Gini 指数,用户应该考虑使用 MAGeCK NEST 来改进hit calling。
5、使用阴性对照序列或非必需基因进行读数归一化
通常需要在单个实验中比较不同条件之间的read-counts。为了在不同生长条件之间进行标准化,理想的标准是靶向所有起始细胞群中完全惰性的基因组位置的sgRNA,这样在任何实验条件下细胞增殖都不会受到不同的影响。AAVS1 是一个经过充分验证的基因座,可用于承载外源基因序列。它具有开放的染色质结构并且具有转录能力。最重要的是,插入或删除 AAVS1 基因座 sgRNA 对细胞没有已知的不利影响,靶向 AAVS1 的 sgRNA 在样本中具有相似的行为(图 3c),表明靶向 AAVS1 的 sgRNA 可能是读取计数的合适对照正常化。使用 AAVS1 靶向 sgRNA 作为对照还可以减轻 Cas9 的核酸酶诱导的毒性并降低整体假阳性率。
与靶向 AAVS1 基因座的 sgRNA 类似,靶向非必需基因的 sgRNA 也可用于标准化读取计数。如果 AAVS1 靶向 sgRNA 不可用,我们编制了一份非必需基因列表(补充数据 1),用于 CRISPR 筛选的标准化。从 927 个在多个 CRISPR 筛选中没有实质性影响的非必需基因开始,我们删除了在多个细胞系中低水平表达的基因。我们在癌细胞系百科全书(CCLE)细胞系(补充图 1b)的 98.3%(1,036 个中的 1,019 个)中选择了表达在第 5-100 个百分位(补充图 1a)中的基因。937 个非必需基因中有 350 个通过了这些标准(补充图 1)。这 350 个基因的表达分布在数百种癌细胞系中是一致的(补充数据 1)。这表明靶向这些基因的 sgRNA 是 CRISPR 筛选标准化的合适对照,如果靶向 AAVS1 的 sgRNA 不可用(图 3d)。MAGeCKFlute 还支持使用非必需基因列表中的 sgRNA 进行读取标准化,我们建议至少包括库中的 200 个非必需基因以确保有效的标准化。
6、拷贝数偏差校正
在 CRISPR 筛选中,在目标基因组位点诱导双链断裂的过程会触发 DNA 损伤反应机制,并可能导致细胞周期停滞,尤其是在具有高拷贝数区域的细胞中。当扩增区域包含目标非必需基因时,观察到的 beta 分数通常比预期的更负面(补充图 2a)。β 分数为负表示敲除该基因可能会抑制细胞增殖或导致细胞死亡,从而在基本基因鉴定中引入了假阳性。如果用户提供了相应的拷贝数文件(示例数据的相应拷贝数文件作为补充数据提供),我们将在protocol中提出一种可选方法(步骤 7A(viii))来纠正与拷贝数相关的偏差2)。在这种方法中,基因组拷贝数和观察到的必要性之间的关系是针对每个实验中的每个基因进行定量建模的。然后根据观察到的结果调整拷贝数偏差,为所有受影响的基因生成校正的 beta 分数。该功能已被整合到 MAGeCKFlute 流程中,可以在执行 MAGeCK RRA 或 MLE 时应用。
7、使用必需基因进行 Beta 评分标准化
暴露于不同条件(有或没有药物治疗)的细胞可能具有不同的增殖率。例如,CDK4/6 抑制剂会影响细胞周期并通常会降低细胞增殖。因此,将倍增时间较快的细胞与增殖较慢的细胞进行比较可能会导致hit 识别的偏差,因为基因似乎在增殖较快的细胞群中具有更强的选择性。在比较使用和未使用药物处理的样品时,通常会出现这种情况,因为许多药物会影响细胞增殖。每个基因的“beta 分数”表示基因正在经历的选择类型:正 beta 分数表示正选择,负 beta 分数表示负选择。当不同样品在CRISPR筛选中同时培养时,倍增时间越短的细胞选择周期越多;因此,快速生长细胞中的基因往往会产生更高的绝对 β 分数(补充图 3a)。为了矫正这种偏差,我们生成了一个包含 625 个精选的、高可信度的核心必需基因的列表(补充数据 3),可用于标准化 beta 分数(有关详细信息,请参阅补充方法)。MAGeCKFlute 使用核心必需基因列表(步骤 11B)对基因 beta 评分进行归一化,假设它们在两个样本之间同样负选择,即使两个样本具有不同的基线增殖率。所有基因的 beta 分数都根据这组精制的 625 个必需基因的中值 beta 分数进行标准化。归一化后,两个样本的回归线的斜率和 x 截距分别接近 1 和 0,这表明在全基因组筛选中对必需基因进行归一化使样本之间的 beta 分数具有可比性(补充图 3b)。对于经过某种处理的 CRISPR 筛选,我们建议用户对必需基因进行归一化,以使处理和对照样本之间的 beta 分数具有可比性。
8、细胞处理后的差异hits识别
在使用必需基因进行 Beta 评分标准化后,下一步是通过减去它们的 Beta 评分来确定治疗和对照条件之间的差异hits。此差异 Beta 分数用于识别与治疗相关的筛选hits。可以在 FluteMLE 函数中指定截止值,默认值为微分 Beta 分数的平均值1 s.d.。我们采用了“分位数匹配”方法来稳健地估计 σ,它是 beta 分数 β 的标准差。选择 σ 使得 β 的绝对值的 (1 − p) 经验分位数与先验正态分布 N(0,σ 2) 的 (1 − p/2) 理论分位数相匹配,其中 p 代表β 分数的分位数。当截止值为 1 sd时,p 设置为 0.32, 当截止值为 2sd时,p设置为0.05,分别对应 68% 和 95% 的 beta 分数落在平均值的 1 和 2 sd 内。如果我们将正态分布的理论上分位数写为 Q N (1 − p),将 β 的经验上分位数写为 Q |β| (1 − p),那么 σ 计算公式为:
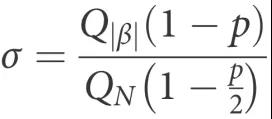
9、筛选hit genes的功能分析
筛选hits的功能分析提供有关在筛选设计中查询的细胞系统生物学的信息。目前广泛使用的功能分析包括 GO 富集分析和 GSEA 分析。正如预期的那样,在简单的增殖筛选中,管家途径的核心成分(例如,核糖体和剪接体)通常被负面选择,并且已发现预测为细胞类型特异性的途径的成分在预测的细胞类型中是必不可少的。
MAGeCKFlute 包含多个功能模块,可用于探索筛选hits的生物学功能。我们包含了从 clusterProfiler、GOstats 和 GSEA 包派生的已发布的富集函数,并添加了富集HGT 以测试基于超几何分布的分子特征的富集。这些功能允许用户指定由 GO 术语、KEGG 通路、MSigDB 基因集集合或用户定义的基因集注释的基因的大小,然后测试它们在筛选hits中的统计过度表现。在某些情况下,用户可能对具有少量基因的蛋白质复合物或通路的强选择感兴趣,因此限制基因集的大小将允许检测到这种富集,而不是被弱选择的大量通路所淹没。
 返回列表
返回列表