


 2020.05.29
2020.05.29  2603次
2603次
小编搜了个25分多的文章来给大家讲讲CRISPR文库筛选耐药基因的思路,由剑桥大学、英国Ralph Lauren乳腺癌研究中心、挪威科技大学和京都大学4家单位共同研究,2020年2月发表在Nature genetics上。
1、文章背景
四分之三的乳腺癌是由雌激素受体(ER,estrogen receptor)调控下诱发,靶向ER通路的药物对大部分ER+疾病妇女有效,但是相当多的女性治疗无效或后期耐药。耐药机制的研究集中在寻找影响ER转录活性的染色质重塑复合物SWI / SNF(最先在酵母中发现这些染色质重塑复合物会导致交配型转换/蔗糖不发酵SWItch mating type/Sucrose Non-Fermenting的表型,简称SWI/SNF)。已经发现有三类常见的SWI / SNF复合物-染色质重塑复合物:BAF(BRG-/BRM-associated factor)、P-BAF和非经典BAF(ncBAF),以及这三类SWI / SNF复合物的核心亚基:BAF(ARID1A,ARID1B,DPF1 / 2/3,SS18)、P-BAF(ARID2,PBRM1,BRD7)和ncBAF(BRD9,GLTSCR1,GLTSCR1L)。为了系统地鉴定与乳腺癌的治疗反应有关的基因功能,研究者采用了全基因组CRISPR文库筛选方法,结合三种不同的治疗方式(两组雌激素受体拮抗剂药物和一组BETi药物),揭示了SWI / SNF复合体是雌激素受体拮抗剂疗效的决定因素。
2、CRISPR筛选显示ARID1A是关键基因
研究者首先构建了全基因组CRISPR筛选文库18,009个人类基因敲除(knock out,KO)的MCF-7乳腺癌细胞,20d时间雌激素受体阻断剂筛选富集耐药的CRISPR的KO库细胞。研究者假定含溴结构域的蛋白质4(BRD4)是ER + 乳腺癌的治疗靶标,用组蛋白乙酰化阅读器靶向含溴结构域和末端外域的抑制剂(BETi,Bromo and extra-C terminal domain inhibitor)作为验证该耐药机制通路的药物,用验证药物同时做CRISPR的KO文库筛选。散点图显示在经过雌激素受体阻断剂筛选26天以后,4个BAF蛋白相关基因比DMSO处理的对照组更多(补充图1)。图1a~c在经过雌激素受体拮抗剂药物筛选26天以后,BAF, P-BAF和ncBAF相关的 sgRNA富集或耗竭情况。图1d~f表明两组ER靶向药物筛选出来的sgRNA基因非常相似,而BET靶向药物(JQ1)筛选出来的sgRNA有明显不同。
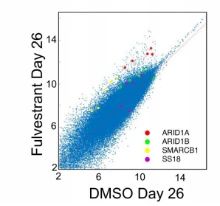
氟维司群(Fulvestrant,雌激素受体拮抗剂, 可通过诱导雌激素受体降解,阻断雌激素受体。

图1a~c Log2差异倍数热图(a)丰度迅速增加的sgRNA:包含已知的ER通路基因;(b)丰度缓慢减少的sgRNA;(c)丰度缓慢增加的sgRNA:包含抑制肿瘤生长的基因。
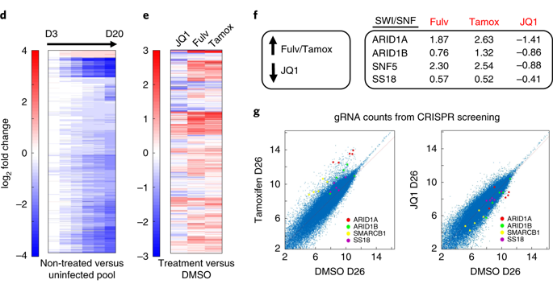
d,层次聚类热图表示感染的第3-20天与增殖有关的sgRNA丰度减少更明显,时间越久,减少越多;
e,层次聚类热图,与DMSO治疗(感染D9后的DMSO对照)相比,两种雌激素阻断剂(氟维司群(Fulv,从300 nM开始并逐渐减少至100 nM),100 nM 4-羟基他莫昔芬(Tamox)或抗癌药BETi(JQ1, 1 µM减少至250 nm)处理26天后基因的log 2倍变化。
f, ARID1A(富含AT的相互作用域1A)和其他BAF的sgRNA在雌激素阻断剂的治疗下,明显富集(数值是这些基因的sgRNA水平相对于DMSO的倍数);而相反的是,在抗癌药BETi治疗下,ARID1A sgRNA丰度明显减少。BAF亚基的ARID1A,ARID1B,SMARCB1和SS18基因的sg丰度在Tamox和JQ1两种药物条件下明显相反。
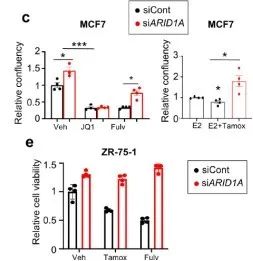
从上面的文库筛选出来的基因中,研究者挑选出在两组ER靶向药物中丰度最高、在BET靶向药物丰度最低的ARID1A继续展开实验。在MCF-7和ZR-75-1细胞中验证了由ER靶向拮抗剂介导的抑癌作用具有ARID1A依赖性。(雌激素受体拮抗剂处理条件下,si ARID1A的细胞生长更快;BETi处理条件下,siARID1A的细胞生长没有差异)(扩展数据图1)
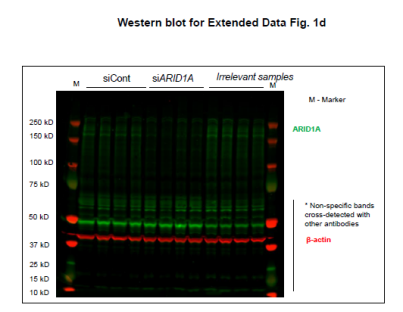
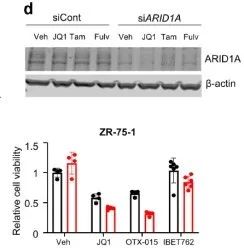
由于siARID1A的细胞增殖能力显著上升,研究者继续构建MCF-7两个单克隆敲除 ARID1A细胞株(克隆11和14)。通过基于Sanger测序证实了ARID1A敲除成功(图2a,补充图4和源数据图2)。
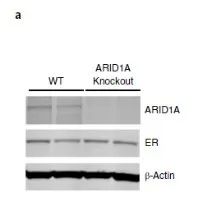
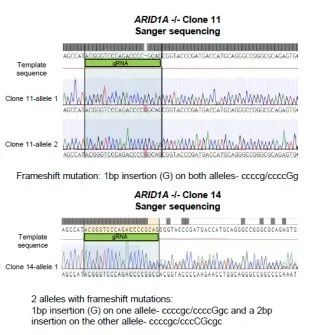
图2a 补充图4

源数据图2
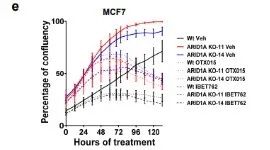
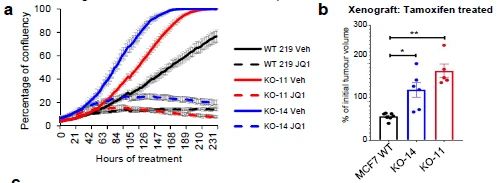
为了排除是脱靶效应导致的细胞增殖变化,研究者用敲除克隆和WT对照的体外生长验证了CRISPR筛选结果,两个ARID1A敲除的细胞克隆增殖能力变强,并且对他莫昔芬有耐药性,但对JQ1表现出敏感性,另外两种临床相关的BET抑制剂(OTX015和IBET762)同样能抑制肿瘤增殖。(图2b和补充图5)。

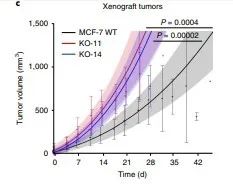
研究者继续做体内实验验证ARID1A敲除克隆促进肿瘤生长,用WT(野生型)、两个ARID1A敲除克隆构建皮下移植肿瘤,持续饲喂雌激素,维持ER + 肿瘤的生长,随后用溶剂或4-羟基他莫昔芬治疗。在4-羟基他莫昔芬治疗下,两个ARID1A敲除的肿瘤比野生型更大(补充图5),这证明抗雌激素必须依赖ARID1A才能发挥功效。然而,在未经治疗的条件下,在ARID1A 敲除的肿瘤明显比WT更大(图2c和补充图5)。),研究者推测他莫昔芬在ARID1A- null肿瘤中的疗效下降可能仅是由于肿瘤增殖能力的增加所致。

3、ARID1A调节ER靶基因,并且是ER复合体的一部分:RNA-seq 表达谱分析
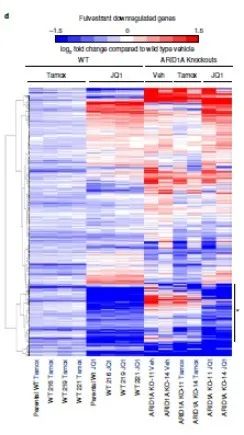
为了探索ARID1A在药物机制中的作用,研究者使用溶剂,氟维司坦,4-羟基他莫昔芬或BETi(JQ1)处理的WT或ARID1A敲除系的四个生物细胞培养样品进行了RNA测序(RNA-seq ),对ARID1A敲除克隆和对照的基因表达分析。不管它们是亲代细胞还是WT克隆系,对照系看起来都相似(补充图8)。

补充图8 图2d
氟维司群和他莫昔芬显示相似的基因表达模式,而JQ1处理则导致基因表达谱有显着差异(图2d和补充图8)。但是JQ1处理ARID1A敲除和野生型的表达谱一样。而在ARID1A敲除细胞中,被氟维司群和他莫昔芬抑制的大多数基因要么上调要么不变(图2d)。总的来说,被氟维司群和他莫昔芬阻抑的86%的基因在ARID1A敲除细胞中不再被显着阻抑,它们(图2d中突出显示)被JQ1处理显着下调至程度与WT细胞相同。因此,即使在没有ER拮抗剂的情况下,ARID1A缺失也导致诱导氟维司群/他莫昔芬抑制的基因的诱导上调,这暗示了ARID1A介导的对ER通路基因的抑制。
研究者用乳腺癌国际联合会(M的ETABRIC)队列研究中ER + 乳腺癌患者数据验证,在溶剂和抗雌激素条件下,ARID1A抑制的基因(在ARID1A基因敲除细胞系中被上调的基因)与在患者中被上调时的不良临床结果相关(图2e和补充图6),再次证明了ARID1A是耐药基因。
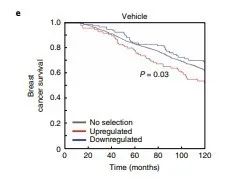
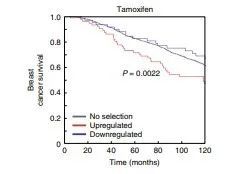
图2e
4、ARID1A有助于HDAC1的募集和乙酰化的调节
研究者通过快速沉淀染内源性蛋白的免疫共沉淀技术(RIME),发现在未经治疗的条件下,组蛋白脱乙酰基酶蛋白HDAC1是与ARID1A相互作用的蛋白(图3a)。
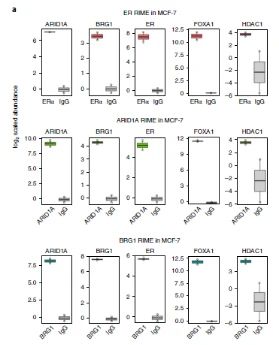
图3a
ARID1A的减少会导致HDAC1结合减少,组蛋白4乙酰化的增加以及在与ER靶向药物在WT环境中通常抑制的基因相邻的调控元件处同时发生BRD4募集(补充图14a),最终导致BET依赖性的肿瘤增殖。

补充图14a
临床结果进一步验证,具有ARID1A突变的乳腺癌患者与具有WT ARID1A的乳腺癌患者显示具有 ARID1A突变型肿瘤的妇女的临床结果较差(图 6d和补充图 14b)。
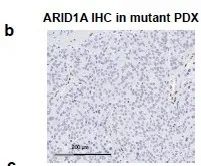
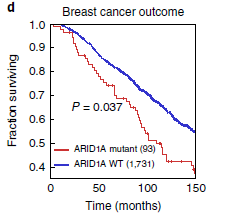
补充图14:ARID1A突变患者呈现ARID1A 阴性 图 6d:ARID1A突变患者生存期更短
取ARID1A 突变女性的PDX肿瘤组织离体培养,经过BETi处理48 h,研究者观察到了增殖指标Ki67的表达减少,说明具有显着的抗肿瘤增殖作用(图6e,f),证实了ARID1A 突变体/缺失型和WT都依赖BET蛋白才能增殖。也提示BETi具有潜在的抗癌药物价值。(比较可惜的是,研究者没有用JQ1治疗解除耐药的实验证据。)
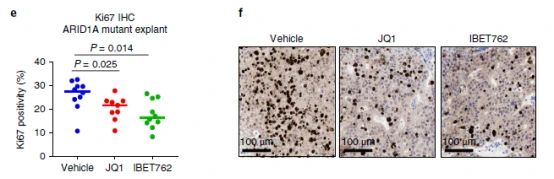
图6e和f
为了进一步验证ARID1A调控细胞增殖的耐药机制,研究者进行了一系列蛋白质组学方法:包括ChIP-seq实验,内源性相互作用的快速免疫沉淀质谱法(RIME),来鉴定ARID1A,BRG1或ER的相互作用,发现ARID1A和两个SWI / SNF常见蛋白BRG1和SNF5(BAF47)的结合位点。SWI / SNF复合物通过先驱因子FOXA1主动结合ER之前被募集到ER 顺式调控元件。ARID1A通过募集HDAC1表现出转录抑制作用,并且当ARID1A功能失活时,会导致HDAC1组蛋白脱乙酰基酶1结合减弱,BRD4组蛋白4赖氨酸乙酰化结合的增加,以及随后的BRD4驱动的转录和肿瘤增殖(图6g)。感兴趣的小伙伴可以查看原文。
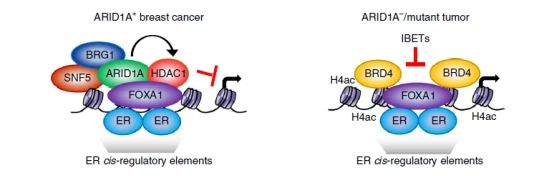
图6g
最后总结一下研究者的思路:首先用全基因组CRISPR文库筛选雌激素受体阻断剂氟维司群和他莫昔芬的耐药靶点。其次用验证耐药靶点的抑制剂BETi(具有潜在的抗癌药物价值)同时做CRISPR文库筛选,筛选出雌激素受体阻断剂中sgRNA高丰度并且BETi筛选下低丰度的ARID1A 和其他3个SWI / SNF复合物亚基是最关键的耐药靶点。接着验证ARID1A失活会导致ER阻断剂抗癌药失效。并用一系列的ChIP-seq实验验证ARID1A和SWI / SNF复合物亚基的转录调控活性。最后用临床证据说明ARID1A突变患者预后更差,针对ARID1A突变患者,可以通过抑制BET通路来减少耐药。
好了,
今天就先解读到这里吧,
祝大家早日发高分文章
 返回列表
返回列表