


 2020.03.09
2020.03.09  1461次
1461次
Aging 2019年的影响因子为5.5,去年一共接收了37篇纯生信分析的文章,在5分以上的杂志中算得上是对纯生信十分友好的了,接下来我们看一个新出炉的案例。

题目:六个特征基因预测头颈部鳞状细胞癌患者生存
方法和结果:从TCGA下载了RNAseq、单核苷酸多态性、拷贝数变异和临床随访数据。根据RNAseq数据和临床随访数据,进行单因素回归分析,筛选到显著与总体生存相关(OS)的基因。根据单核苷酸多态性和拷贝数变异数据筛选到显著变异的基因。将与OS相关的基因和变异基因取交集,然后再用随机森林进一步筛选,最终筛选到6个基因。将TCGA样本分为训练集和测试集,用这6个基因作为特征,用训练集建立预后模型,结果发现模型能很好地预测预后(P
结论:构建了一个6基因特征作为预测HN SCC患者生存的一个新的预后指标。
这篇文章分析亮点有两个:
1.筛选特征基因的方法,抛弃了传统的差异表达分析,而是利用回归分析、变异分析、随机森林三步筛选,最终确定了特征基因;
2.多次验证,本文在建立预测模型之后的所有验证工作,都在三个不同数据集中进行了验证,确保结果的稳定性,更有说服力。
下面,我们来看看详细的过程吧~
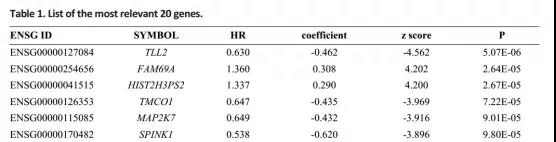
1、鉴定与OS相关的基因
单因素Cox回归分析TCGA头颈部鳞状细胞癌患者的RNA表达数据,筛选P
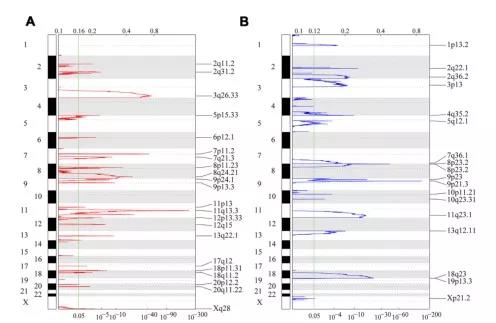
2、识别显著变异的基因
用GISTIC 2.0分析TCGA拷贝数变异数据,以识别显著扩增或缺失的基因。一共筛选到247个显著扩增基因和901个显著缺失的基因。AB分别表示了各个染色体上拷贝数扩增、缺失的情况。
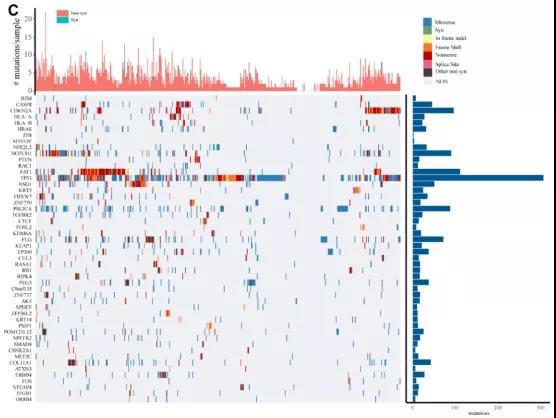
用Mutsig2分析了TCGA突变注释数据,以识别显著高频突变的基因。本次分析筛选了302个高频突变的基因。C图展示了各个基因的不同突变在各个样本的情况,突变种类包括:同义突变、错义突变、插入或缺失、移位、无意义突变、剪切位点分布和其他非同义突变。从图中可以看出,不同的基因发生的突变的种类有很大的不同,但是相同的是,这些基因的突变可能和肿瘤的发生和发展都有一定的关联。
3、变异基因的功能分析
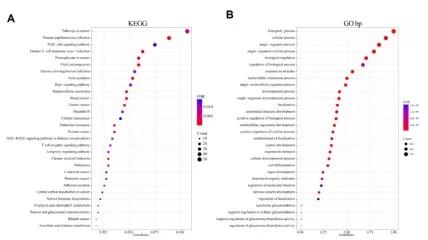
将GISTIC 2.0和Mutsig2分析得到的显著突变的基因取交集,结果得到1321个基因。对这些基因做GO和KEGG富集分析。KEGG富集分析结果显示,这些基因显著富集在癌症、HPV感染、PI3K-Akt信号通路、人T细胞白血病病毒1感染、人巨细胞病毒感染以及许多其他与癌症的发展相关的通路(图A)。GO富集分析显示,这些基因显著富集在发育过程、细胞过程正调控、细胞分化和定位调控等通路(图2B)。这些通路也与癌症的发生和发展密切相关,也就是说,表现出基因组变异的基因与癌症密切相关。
4、特征基因筛选
将筛选的1321个基因组变异基因和预后相关的425个基因取交集,共得到36个候选基因。然后用随机森林作进一步的特征筛选,利用误差率与分类树数之间的关系,以相对重要性大于0.4的基因作为终止信号,最终筛选到6个特征基因。
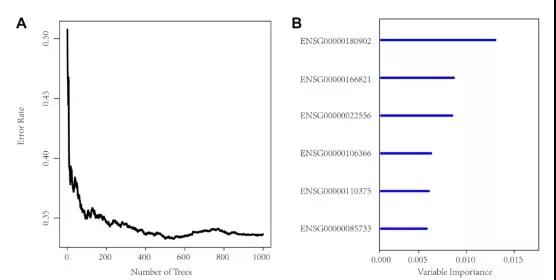
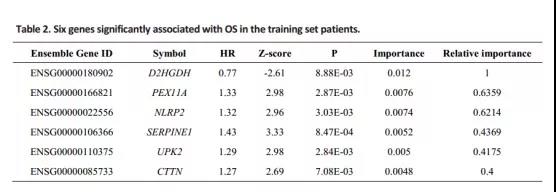
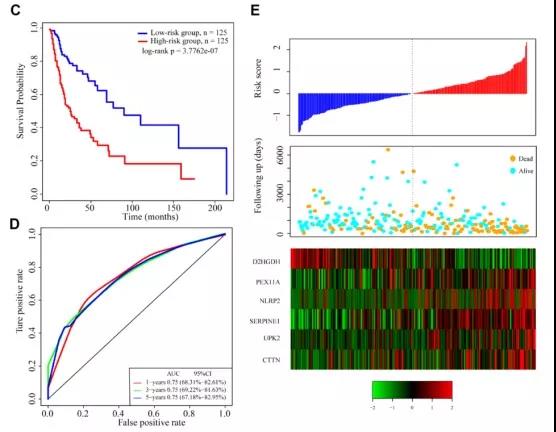
再将TCGA病人分为训练集和测试集。首先根据训练集样本这6个基因的表达量、单因素Cox回归系数,建立如下的风险评分模型。

计算训练集每个样本的Risk分数,然后以Risk分数的中位数将样本分为两类:Risk分数大于中位数的样本被定为高风险,低于中位数的样本被定为低风险。将高风险病人和低风险病人进行生存分析比较,发现高风险病人预后更差(图CE)。6基因特征的1年、3年和5年平均AUC为0.75(图D)。
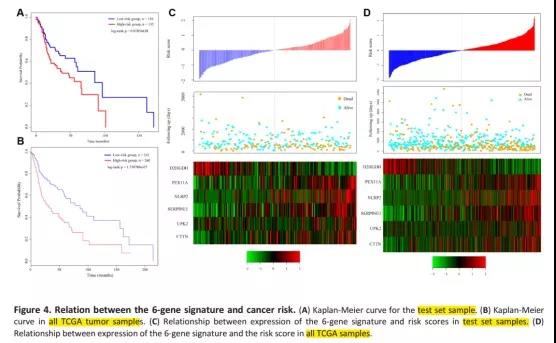
5、预后模型验证
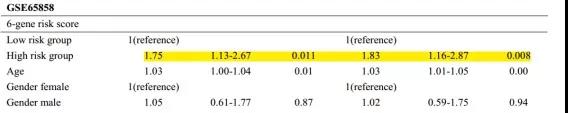
文章用TCGA测试集、TCGA所有数据集和GSE65858数据集分别对预后模型进行验证。都得到了与训练集相似的结果(高风险组预后较差)。
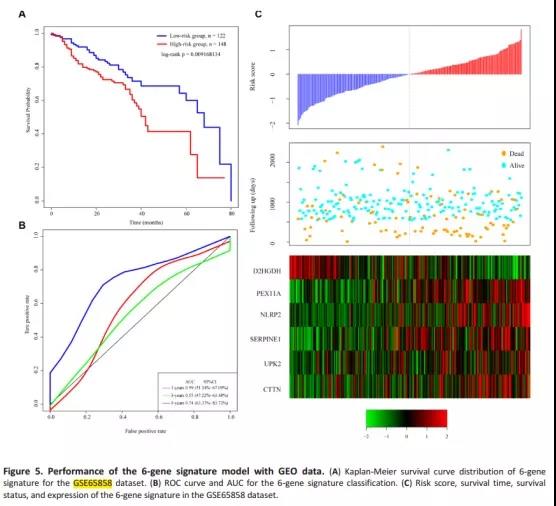
6、预后模型的临床独立性分析
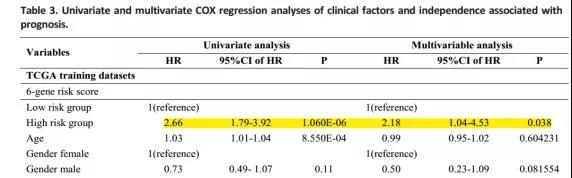
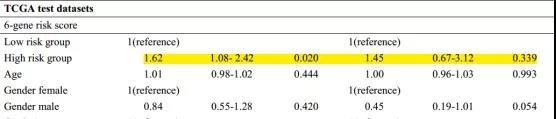
将建立的预后模型与其他临床数据(年龄、性别、TMN分期和肿瘤分级)进行单因显示,该预测模型中的6个基因是一种独立于其他临床因素的预后指标,在临床应用时表现出独立的预测性能。
7、GSEA(基因富集分析)高风险和低风险人群路径差异
在TCGA训练集中使用GSEA分析高风险和低风险人群中显著富集的途径。分析发现:有20条显著富集的通路,其中局灶性粘附、TGF-β信号通路、WNT信号通路和ERBB信号通路与肿瘤的发生、发展和转移密切相关,并且这些途径在高风险样本中显著富集。
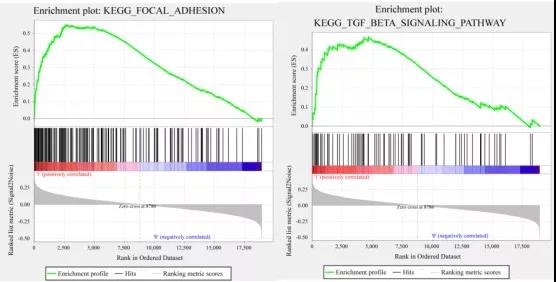
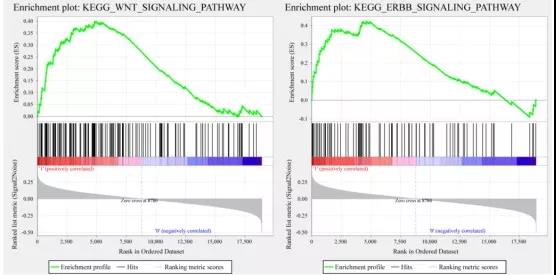
这篇文章不用做实验,所有用到的突变、RNAseq、临床跟踪数据都可以在TCGA、GEO数据库中下载到。由此可见,通过对数据进行灵活挖掘运用,一样可以发高分文章!
 返回列表
返回列表