


 2021.11.15
2021.11.15  1931次
1931次
一、导言
小编今天给大家分享一篇2021年5月发表于Nature的关于肿瘤免疫治疗的文章——TIM-3 restrains anti-tumour immunity by regulating inflammasome activation.肿瘤免疫治疗是现在乃至未来研究的一个大热点,而T细胞免疫球蛋白粘蛋白3(TIM-3)是继PD-1/PD-L1和CTLA-4之后,又一个新兴的免疫检查点分子。TIM-3在肿瘤微环境中CD8+ T细胞上的表达被认为是T细胞功能障碍的主要标志,然而,TIM-3也在其他几种类型的免疫细胞上表达,那么T细胞之外的免疫细胞很有可能参与抗肿瘤作用。在这里,研究者采用敲除TIM-3和单细胞RNA测序证明了TIM-3对树突状细胞(DCs)的重要性,即DCs上TIM-3的缺失——而不是CD4+或CD8+ T细胞,促进更强大的抗肿瘤免疫作用。TIM-3的缺失使DCs无法表达调节程序,并促进了CD8+效应和干细胞样T细胞的维持。此外,DCs中TIM-3的缺失导致活性氧簇的积累增加,从而导致NLRP3的激活。抑制恶性肿瘤组织激活或下游表达的细胞因子IL-1β和IL-18,完全消除了DCs中TIM-3缺失所观察到的保护性抗肿瘤免疫。研究者揭示了TIM-3在调节DC功能中的重要作用,并强调了阻断TIM-3通过调节炎症小体的激活来促进抗肿瘤免疫的潜力。
二、结果展示
1、树突状细胞上缺失TIM-3可以减少肿瘤负担
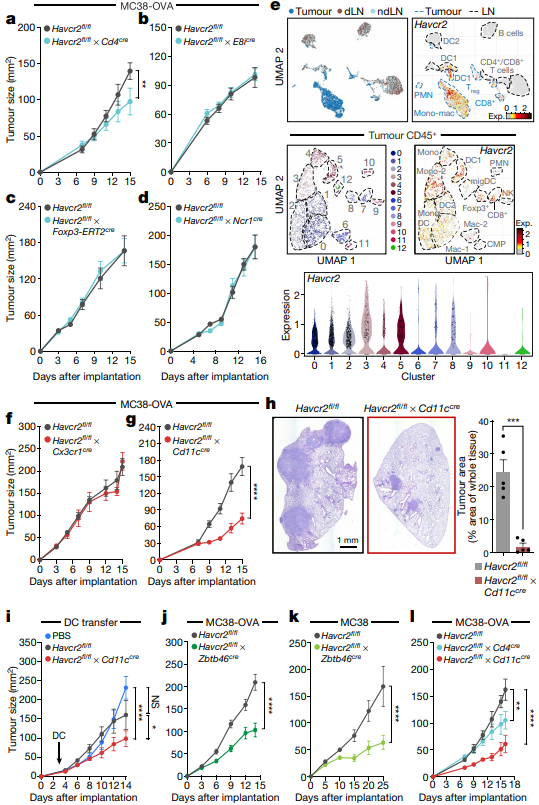
为了探究TIM-3在T细胞中的功能,研究者制备了条件性敲除小鼠,敲除 CD4+和CD8+ T细胞、CD8+ T细胞或调节性T(Treg)细胞中的TIM-3。结果发现,CD4+和CD8+ T细胞的缺失可适度减少免疫原性MC38结肠癌的肿瘤负担,而CD8+ T细胞或Treg细胞的缺失则没有影响。TIM-3在自然杀伤细胞(NK)上也有表达,但是,使用Ncr1cre特异性缺失对肿瘤生长没有影响。这些意外的发现,使研究者认为TIM-3在调节抗肿瘤免疫中的作用,可能主要是通过髓系细胞,特别是DCs介导的,其TIM-3在体内高表达。最近,通过对人类疾病的观察,髓系细胞中TIM-3表达的重要性变得明显,编码TIM-3的Havcr2基因的突变,与髓系细胞的过度激活和IL-18水平的升高有关。
为了全面探究TIM-3的表达谱,研究者对肿瘤浸润CD45+细胞(肿瘤浸润淋巴细胞(TILs))进行了单细胞RNA测序分析(scRNA-seq),并对野生型 MC38-OVAdim肿瘤小鼠引流和非引流淋巴结中的CD45+细胞进行测序比较。结果发现,在淋巴组织中,Havcr2的表达在表达经典DC1标志物Xcr1和Clec9a的一簇细胞中最为突出。然而,在肿瘤中,Havcr2表达于多种细胞类型,包括DCs、CD8+ T细胞、Treg细胞、单核细胞和巨噬细胞。使用Cd11ccre敲除DCs中的TIM-3,观察到肿瘤生长显著减少。进一步用 KP1.9肺腺癌细胞株进行验证,发现Havcr2cko小鼠肿瘤负担显著减少。DC-transfer实验也证明了树突状细胞内在的 TIM-3的缺失足以促进抗肿瘤免疫。研究者还比较了临床上树突状细胞和T细胞上TIM-3的缺失情况,发现DC人群中TIM-3的缺失导致更好的肿瘤生长抑制。
2、TIM-3缺失的DCs扩增干细胞样CD8+T细胞
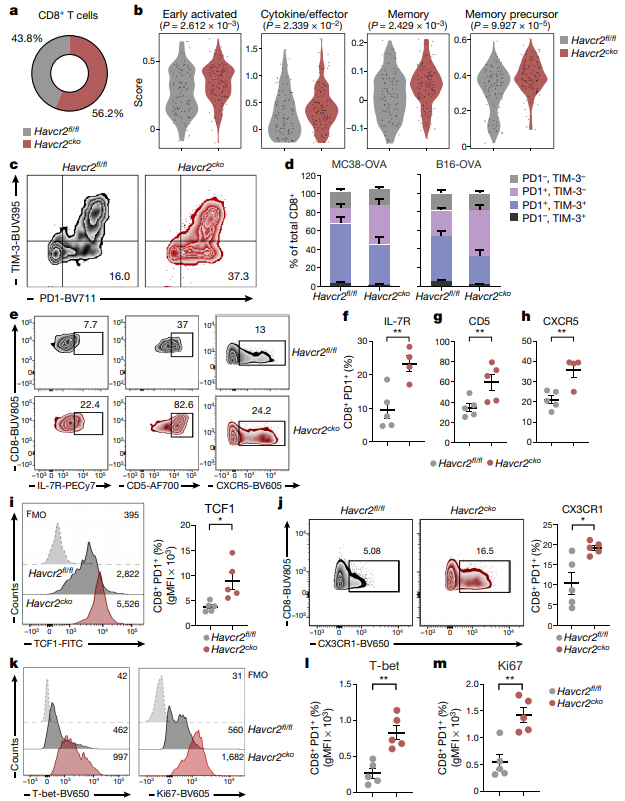
利用scRNA-seq,研究者分析了Havcr2fl/fl和Havcr2cko小鼠中携带 MC38-OVAdim 肿瘤的TILs,鉴定了15种不同的细胞类型,包括4个淋巴样细胞群,1个NK细胞群,7个不同的髓样细胞群和3个cDC群。在Havcr2fl/fl和Havcr2cko小鼠的样本中,所有的簇都有表达,但是比例不同。特别是集群0(包含单核细胞衍生的巨噬细胞)、集群10和12(包含活化的B细胞)和集群7(包含CD8+T细胞)在Havcr2cko小鼠的肿瘤中表现更为突出。
通过分析CD8+T细胞(集群7),观察到Havcr2cko小鼠的肿瘤细胞表达更高水平的基因特征,这些基因特征存在于一系列功能状态中,包括早期激活、效应、记忆和记忆前体。研究者假设树突状细胞缺失TIM-3可能有助于诱导或维持干细胞样CD8+T细胞,观察到Havcr2cko小鼠的肿瘤中PD-1单阳性TILs 的频率和绝对数量显著增加可支持这一观点。PD-1+ TILs已被证明是包含干细胞样和效应样CD8+TILs。因此,研究者评估了细胞表面标记物的表达,区分这两个种群。研究发现,在Havcr2cko小鼠中浸润肿瘤的PD-1+ CD8+T细胞上IL-7R、CD5和CXCR5的表达增加,表明干细胞样CD8+TILs数量增加。与这些结果相一致,在Havcr2cko小鼠的肿瘤中观察到CXCR5+ TIM-3-CD8+T细胞的显著扩增,以及TCF1(记忆前体T细胞的特征)。研究者进一步发现CX3CR1、T-bet和ki67的表达增加,表明进入效应谱系的细胞数量增加。这些结果表明,DCs上TIM-3表达的缺失促进了干细胞样CD8+T细胞和效应T细胞的维持,共同促进了Havcr2cko小鼠肿瘤的保护性免疫应答。
3、TIM-3缺失增强抗原特异性免疫
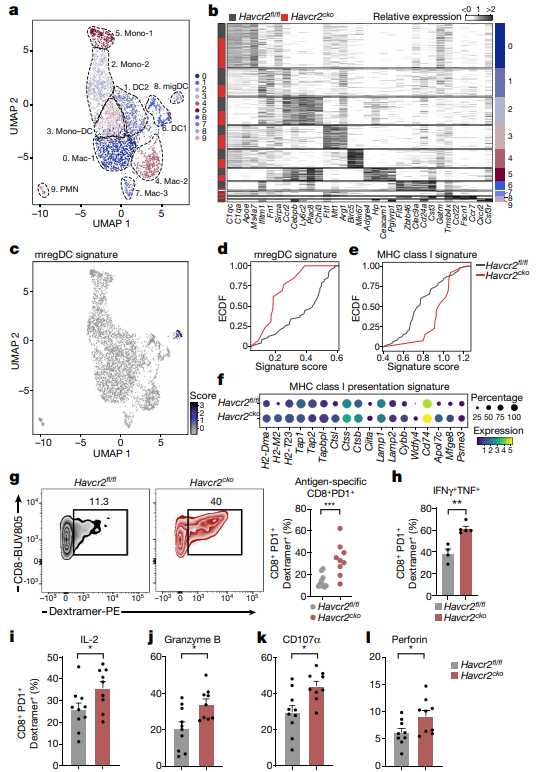
通过对单细胞数据集中的髓系细胞进行亚群分析,发现10个不同转录的亚群,这些亚群在Havcr2fl/fl和Havcr2cko小鼠的肿瘤中都有表达,但比例不同。特别是集群0(CCR5表达单核细胞衍生的巨噬细胞)在Havcr2cko小鼠的肿瘤中富集。相反,在集群9(嗜中性粒细胞)、集群6(DC1s)和集群8(migDCs)中的比例较小。
migDC群已经被证明在肿瘤微环境中获得调节程序,导致成熟的树突状细胞富含免疫调节分子(mregDCs),抑制了这些细胞的免疫原性潜力。在髓细胞上得到这个特征确实突出了簇8,migDCs在Havcr2cko小鼠中的评分较低,伴随着免疫调节分子在 mregDCs中的表达减少,包括IL-4R、CD200、CD83和OX-40。研究者进一步探究IL-4阻断剂对Havcr2cko荷瘤小鼠的影响。结果发现,抗IL-4治疗可明显延缓Havcr2fl/fl小鼠的肿瘤生长,但对Havcr2cko小鼠无效。这说明 TIM-3的缺失可能会阻碍mcreDC程序的获得,使migDCs能够获得完整的免疫原性程序。通过对髓样细胞群进行评分,发现Havcr2cko集群8 migDCs有明显富集,其中与MHC I类主要组织相容性复合体相关的几个基因,包括Wdfy4、Cybb、 Tapbpl和H2-M2被上调。体外细胞实验发现,Havcr2cko小鼠肿瘤中抗原特异性CD8+T细胞的频率增加,产生更高水平的效应细胞因子IFN-γ、TNF和IL-2,具有更大的细胞毒性潜能,增加颗粒B、CD107α和穿孔素的表达,导致体内CD8+T细胞溶解活性的增强。
4、缺失TIM-3促进炎症激活
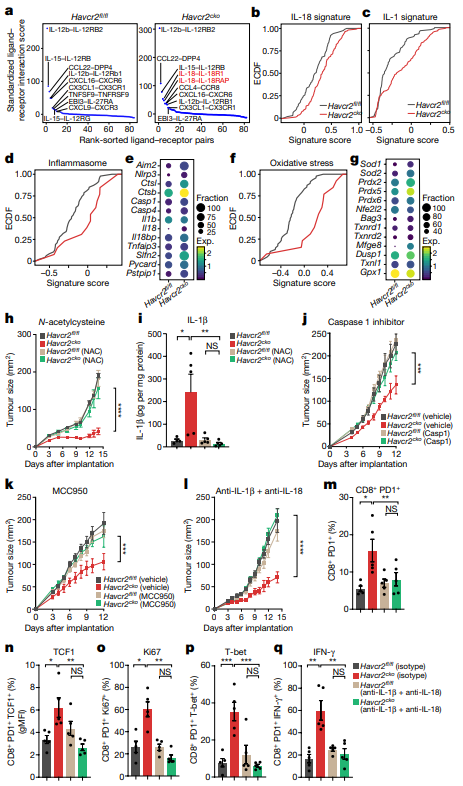
通过进一步的生物信息学分析和细胞因子分析显示,Havcr2fl/fl和Havcr2cko 小鼠的肿瘤组织中IL-1β和IL-18表达水平较高。IL-1β和IL-18都是紧密调节的细胞因子,在炎症介导的过程中作为成熟蛋白释放。通过对Havcr2fl/fl和Havcr2cko小鼠炎性基因差异表达的研究,发现Havcr2cko migDCs的炎性基因明显富集,包括涉及炎性基因激活的上游(Aim2和Nlrp3)和下游(Casp1、Casp4和IL18)接头分子。
研究者假设,内源性损伤相关的分子模式,包括活性氧类、氧化脂质和氧化 DNA,可能在肿瘤浸润的Havcr2cko DCs 中引起炎症组激活。事实上,Havcr2cko migDCs在与氧化应激相关的基因特征上得分更高,其关键酶SOD1、SOD2、BAG3和PRDX5的表达上调。另外检测了MC38-OVAdim肿瘤的DCs的活性氧类活性,发现Havcr2cko DC1s和migDCs的活性显著增加。此外,抗氧化剂N-乙酰半胱氨酸可以完全逆转对Havcr2cko小鼠肿瘤生长的保护性抑制。减少 Havcr2cko小鼠肿瘤中的氧化应激也能完全消除IL-1β的分泌。
最后,研究者采用3种不同的方法在体内靶向炎症途径:(1)caspase 1抑制剂干扰 pro-IL-1β和pro-IL-18分裂;(2)MCC950干扰炎症小体复合蛋白ASC寡聚化;(3)抗IL-1β和抗IL-18阻断抗体(anti-IL-1β/IL-18)抑制下游细胞因子。每种方法都可以消除Havcr2cko的保护性抗肿瘤免疫作用,其中anti-IL-1β/ IL-18的效果最强。此外,流式细胞术分析治疗的Havcr2cko小鼠肿瘤显示完全逆转的CD8+表型。值得注意的是TIM-3被证明是通过与TIR8相互作用,负向调节IL-1β和IL-18受体信号。这种相互作用在DCs中是如何发挥作用的,目前尚不清楚,但可能牵涉到一种自分泌前馈环路。在这种环路中,缺失DCs上的TIM-3会导致IL-1β的生成,同时增强IL-1β的反应,共同促进免疫原性DCs的产生。
三、总结
TIM-3已经成为一个极具吸引力的治疗靶点,新出现的数据表明,在AML和骨髓增生异常综合征中,TIM-3的阻断会诱发50-60%的应答率,这表明TIM-3的阻断可以作为髓系肿瘤的治疗策略,而其他检查点抑制剂的治疗效果则有限。该文章证明了TIM-3的缺失,阻碍migDCs 获得调节程序,促进由IL-1β和IL-18驱动的CD8+T细胞效应的维持。这些结果进一步证明了通过刺激炎症通路治疗癌症的可行性,并强调了阻断TIM-3在释放炎症通路以增强抗肿瘤免疫力方面的价值。
 返回列表
返回列表